Abstract
A genetic lab investigation to disarm the trpC enzyme in bacteria – Escherichia coli to confirm recombination for kanamycin resistance. Its principal aim is attaining the best probability of success. According to one theory, there may be around 500 or 700 base pairs. The core objective should be addressed through further testing and research.
Introduction
The trpC gene in E. coli is removed through recombination and replaced by kanamycin during investigation (Murphy, 2016). E. coli involves focusing on TrpC, a vital enzyme. The goal was to prevent the enzyme in an important gene alteration process, put kanamycin instead of trpC, and make recombination occur (Murphy, 2016).
All possibilities involving the trpC gene of E. coli (Escherichia coli) must be considered TrpC, one of the key enzymes in E. coli (Poteete, 2008). Replacement of trpC by kanamycin will create multiple cycles of denaturation, extension, and annealing of the enzyme. Several methods, however, will be used during investigations for quick rebinding in the trpC-kanamycin case (Khodursky et al., 2000). The other hypothesis proposes that there could have been as much as a thousand-six hundred base pairs following numerous annealing, denaturing, and extending cycles. A more speculative theory may be the development of kanamycin resistance (Wong QN et al., 2005).
Materials and Methods
It involved placing 1 gram of Agarose powder in a microwave for 30 seconds to make a 50 ml Agarose gel solution, which was measured using a graduated cylinder. After that, they carefully added 0.5 milliliters of DNA gel stain into the mixture using a pipette pre-calibrated for 0.5-10 microliters. Then, the mixture was placed in the gel chamber and allowed to set. It entailed mildly pouring buffered fluid on the solidified gel to conduct the electrophoresis comb. Loading started with 7 mL of the ladder introduced in the first slot and 5 mL dye loading in the appropriate PCR slot. Wong QN et al. (2005) also employed a DNA centrifuge purification column to centrifuge a mixture containing 200 microliters of water, 100 ml of binding buffer, and 45 ml of PCR fragments.
The DNA had to be prepared carefully for effective amplification through the PCR apparatus. As per Mienda BS et al. (2015), precise amounts of essential components were mixed: Thermopolis buffer – five milliliters; deoxyribonucleotides triphosphates – three milliliters; magnesium sulfate – one milliliter; template DNA – one milliliter; and each primer – one milliliter. These constituents were carefully mixed in accurate amounts, adding up to 50 microliters, which was imperative for the PCR procedure. Adding half a microliter of polymerase accelerates the amplicon. This specially prepared mixture is placed into a thermocycler, which performs successive heat cycles of denaturation, annealing, and extension required for DNA replication and amplification.
Handle the second agarose gel carefully for electrophoresis once the first one has been done. Run electrophoresis for 40 minutes for the PCR mixture loading at 90 and 120 amps upon loading. Every half an hour, conducting a preliminary assessment of compliance with the plan for the separation process would be necessary. If you want the best results, increase the electrophoresis time for another 10 minutes. For instance, statistics showed up to 500-700 pairs in the trial group. The GeneRuler 1 kb Plus DNA Ladder was identified at 500 base pairs by Khodursky et al. (2000), and it has become a reference point for further studies on this issue. Poteete’s 2013 study provided important information on expected fragment sizes and, hence, the usefulness of experimental procedures and gene amplicon within the trpC gene region—efficient and quick testing involved Overlap Extension PCR plating.
Poteete (2013) states that competent cells successfully uptake DNA fragments when appropriately distributed with two microliters of PCR product. The DNA was introduced into the cells in a Bio-Rad 165-2086 cuvette by four-second electroporation at 2.0 KV. One milliliter of the LB broth was added into a tube and warmed at 37°C before shocking the cells. This procedure allowed the cells to recover and operate for about thirty minutes. This process comprised a uniform coating of plate components and the commencement of bacterial growth for colony observation.
Results
The gel electrophoresis data from Figure 1 indicated that the experimental group fragments ranged from 500 to 700 base pairs. However, the GeneRuler 1kbp Plus DNA Lader only had fragments for 500 base pairs in row 1.
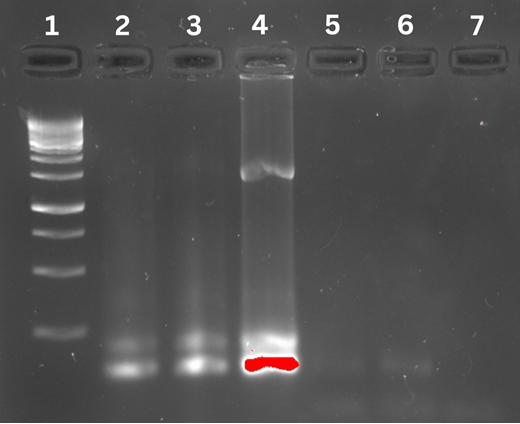
Figure 1 shows the results of gel electrophoresis and Kanamycin Cassette analysis conducted on the Plasmid type per Murphy’s paper dated 2016. The first row shows the GeneRuler 1 kb Plus DNA Ladder, and the experimental 3 is in the second row.
After pouring 100 microliters of electroporated competent cells and 2 microliters of PCR product from cuvettes 1 and 2, both samples showed growth on all LB agar plates. Three colonies were observed on the surface of three LB agar plates and another three LB agar plates with kanamycin, as mentioned in Figure 2. However, no growth was observed on LB and kanamycin-containing agar among the samples.
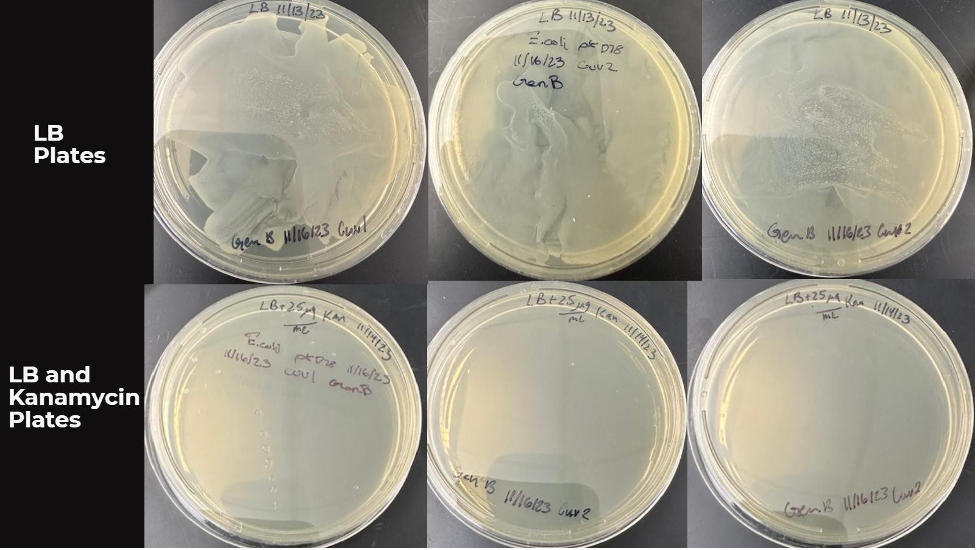
Findings from the observations on LB and LB Kanamycin plate cultures are shown in Figure 2. Wang X et al. (2020) observed that these involved both PCR products and the culture result from electroporated competent cells. Row 1 represents Cuvette 1, whereas rows 2 and 3 are for Cuvette 2.
Discussion
The primary goal was to test for kanamycin resistance after recombination had successfully replaced the trpC gene with a kan cassette (Poteete, 2009). According to Datsenko et al. (2000), the trip PCR result at 500-700 base pairs takes much less time to be detected by gel electrophoresis than anticipated. Problems may arise if there is a difference of 780 base pairs from the expected 1600 base pairs to effectively integrate the Kan cassette (Vandierendonck et al., 2023). Satisfactory Datsenko et al. (2000) found no development when electroporated cells containing PCR items were plated on LB and kanamycin plates, recommending an awkwardness within the chemical arrangement or insufficient quality loosening up. This proves that the endeavor to form bacteria resistant to anti-microbials fizzled.
According to Poteete (2008), the foremost plausible causes incorporate heresy or lack of kanamycin consolidated into the quality. These blended raise questions about the fundamental thought that interfaces kanamycin-induced quality erasure to the advancement of anti-microbial resistance and highlight the need for more trials to affirm the comes about (Vandierendonck et al., 2023). Concurring with Khodursky et al. (2000), there may have been issues with appropriately supplanting kanamycin due to uneven arrangement chemistry, deficient quality loosening up, or lack of test input on the agarose gel. Investigating how the E. coli tryptophan digestion system is influenced by trpC gene deletion is critically required, as highlighted by Hoppel et al. (2022).
There may be a criticism circle between the misfortune of indole-3-glycerophosphate synthetase (IGPS) and the work of tryptophan amalgamation, which impacts tryptophan levels. The part of the quality taking after erasure can be discovered by examining the tryptophan digestion system. The cancellation influences the coordinated pathway, highlighting the results of disturbed trip quality movement or nonappearance. Extra inquiry about the tryptophan digestion system in E. coli is required to demonstrate this. Still, future considerations ought to approve the recombination handle, assess the viability of quality expulsion, and explore issues like quality releasing, blasphemy, or wasteful kanamycin consolidation. sWhen the trpC gene is deleted from O. trpC, metabolic and physiological processes are more severely affected.
Conclusion
The main objective of this study was to replace the primary enzyme of E. coli with the kanaymic cassette, expecting it to replicate and resist kanamycin. Nevertheless, the experiment had challenges because of few replication attempts and antibodies. Consequently, there was a shorter PCR product than anticipated. Analysis of the Trip PCR products by gel electrophoresis indicates that there were approximately 500-700 pairings instead of the expected amount. Following the plating of live cells and PCR products on LB and kanamycin plates, no growth was revealed, suggesting reactivation and mutations of antibodies.
The discovery challenges the suggested link between gene deletion resulting from kanamycin treatment and drug resistance. This underlines the need for more in-depth investigation. These observed differences may be due to the absence of good gene unwinding, infection, or kanamycin’s influence over genes. These complex issues can be investigated; thus, future research efforts should focus on unveiling the specific impacts of trpC.
Summary
This work aims to substitute the primary enzyme of E. coli with a cassette that includes kanamycin to accomplish both reproduction and kanamycin resistance. The PCR outcome was of reduced length compared to the anticipated result, necessitating the utilization of the necessary antibody in the experiment. There were anticipated 500-700 correlations between trip PCR results and gel electrophoresis. The attempt to culture live cells and plate PCR products on LB and kanamycin media was unsuccessful in generating colonies due to reactivation and alterations in antibody composition. Although these data do not establish a direct cause-and-effect relationship between medication resistance and kanamycin-induced gene deletion, they indicate the need for additional inquiry. The identified mutations may have been produced by insufficient gene unwinding, infection, or the impact of kanamycin on the gene. Future research should prioritize quantifying the effects of journeys.
References
Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proceedings of the National Academy of Sciences. 97(12).
Khodursky A.B., et al. 2000. DNA microarray analysis of gene expression in response to physiological and genetic changes affecting Escherichia coli’s tryptophan metabolism.
Mienda BS, Shamsir, MS. 2015. Model-driven in Silico glpC Gene Knockout Predicts Increased Succinate Production from Glycerol in Escherichia coli. AIMS Bioengineering 2(2):40-48.
Murphy KC. (2016). Lambda Recombination and Recombineering. EcoSal Plus. 7(1).
Poteete AR. (2008). Involvement of DNA replication in phage lambda Red-mediated homologous recombination. Mol Microbiol 68:66–74
Poteete AR. (2009). Expansion of a chromosomal repeat in Escherichia coli: roles of replication, repair, and recombination functions. BMC Mol Biol 10(14). doi:10.1186/1471-2199-10-14.
Poteete AR. (2013). Involvement of Escherichia coli DNA Replication Proteins in Phage Lambda Red-Mediated Homologous Recombination. Plos One 8(6).
Schoppel, K, Trachtmann, N, Korzin, EJ, et al. 2022. Metabolic control analysis enables rational improvement of E. coli L-tryptophan producers, but methylglyoxal formation limits glycerol-based production. Microb Cell Fact 21:201
Vandierendonck J, Girardin Y, De Bruyn PD, et al. 2023. A Multi-Layer-Controlled Strategy for Cloning and Expression of Toxin Genes in Escherichia coli. Toxins. 15(508).
Wang X, Policarpio L, Prajapati D, Li Z, Zhang H. 2020. We are developing E. coli-E. Coli co-cultures to overcome barriers of heterologous tryptamine biosynthesis. Metabolic Engineering Communications. 10.
Wong QN, Ng VC, Lin MC, Kung HF, Chan D, Huang JD. 2005. Efficient and seamless DNA recombineering using a thymidylate synthase A selection system in Escherichia coli Nucleic Acids Res 33(59).
 write
write